Fda Legal Manufacturer Definition
Manufacturers (domestic and foreign) and primary distributors (importers) of medical devices must register their facilities with the FDA. All company registrations must be filed electronically, unless the FDA has granted an exemption. All registration information must be reviewed annually between October 1 and December 31 of each year. In addition to registration, foreign manufacturers must also appoint a U.S. representative. From 1 October 2007, most establishments will have to pay a registration fee. u) Manufacturing means all stages of propagation or manufacture and preparation of products and includes, but is not limited to, bottling, testing, labelling, packaging and storage by the manufacturer. (x) %quot%batch%quot% means the quantity of uniform material which, in accordance with the manufacturer`s instructions, has been thoroughly mixed in a single container. (ii) the applicant is the manufacturer of a significant percentage of the blood supply in the United States; Significant interruption means a change in production that is reasonably likely to result in a reduction in the supply of an organic product by a manufacturer that is more than negligible and that affects the manufacturer`s ability to fulfill orders or meet the expected demand for its product and that does not result in production interruptions due to issues such as routine maintenance or insignificant changes in production. Production as long as the manufacturer expects a recovery.
Operations in a short time. Repacker – Packs finished equipment in bulk or packs a manufacturer`s equipment in various containers (excluding shipping containers). (1) When used, the terms “manufacturer”, “repackager”, “relabeler” or “re-consignor” means a manufacturer, repackager, relabeller or reclaimer located in a foreign country that manufactures, repackages, relabels or saves a drug or feed that contains or contains a new veterinary drug that is imported or offered for importation into the United States. Serious adverse experiences. Any adverse experience that occurs at any dose and results in any of the following: death, life-threatening adverse experience, hospitalization or extension of an existing hospitalization, persistent or significant disability or disability, or congenital anomaly/congenital anomaly. Major medical events that may not result in death, be life-threatening, or require hospitalization may be considered a serious adverse experience if, in reasonable medical judgment, they may endanger the patient or subject and may require medical or surgical intervention to prevent any of the outcomes listed in this definition. Examples of such medical events include allergic bronchospasm that requires intensive treatment in an emergency room or at home, blood dyscrasias or seizures that do not result in hospitalization, or the development of substance abuse or substance abuse. (t) %quot%quality audit%quot% means a systematic and independent examination of a manufacturer`s quality system, carried out at specified intervals and with sufficient frequency to verify that both the activities of the quality system and the results of those activities comply with the procedures of the quality system, whether those procedures are applied effectively and whether those procedures are appropriate for achieving the objectives of the quality system: Quality assurance system. U.S.
Export Equipment Manufacturer Only – Manufactures medical devices that are not sold in the United States and are manufactured exclusively for export abroad. (e) records in case of shared manufacturing responsibility. Where two or more establishments are involved in the manufacture of a product, the accounts kept by each establishment must clearly indicate the extent of its responsibility. In addition, each participating manufacturer shall provide the manufacturer preparing the product in its final form for sale, exchange or exchange with a copy of all records of the manufacturing operations carried out by that participating manufacturer with regard to the safety, purity and efficacy of the batches of the product concerned and the manufacturer who reprocesses the product in its final form: keep complete records of all manufacturing operations related to the product. (j) Patient Privacy. For biologics other than vaccines, the applicant shall not include the names and addresses of individual patients in the reports referred to in this Section; Instead, the applicant should assign a unique code to identify the patient. The applicant must provide the name of the notifier from whom the information was received as part of the Foreground Information, even if it is the patient. The names of patients, healthcare professionals, hospitals, and geographic identifiers in adverse experience reports may not be disclosed to the public in accordance with FDA public information regulations in Part 20 of this chapter. For reports of adverse experiences with vaccines, this data is part of the CDC Privacy Act system 09-20-0136, “Epidemiological Studies and Surveillance of Disease Problems.” Information identifying the person who received the vaccine or their legal representative will not be made available to the public, but may be made available to the vaccinated person or legal guardian. For the purposes of registration and registration under this Part, when used to change the term “owner”, “manufacturer”, “repackager”, “relabeler”, “reseller”, “private label distributor” or “establishment” means a registrant, manufacturer, repackager, relabeler, distributor or private label entity in a state or territory of the United States, the District of Columbia or the Commonwealth of Puerto Rico.
Manufacturer – Manufactures, by chemical, physical, biological or other processes, any article that meets the definition of “device” in Section 201(h) of the Federal Food, Drug and Cosmetic (FD&C) Act. (v) Location includes all buildings, fixtures, equipment and animals used, as well as personnel employed by a manufacturer in a specified area identified by an address appropriate for identification. (hh) Distributed means that the biologic has left the control of the authorized manufacturer. (g) wholesaler: any person engaged in the wholesale distribution of medicinal products subject to medical prescription, including, but not limited to, manufacturers; packer; private label retailers; private label dealers; Worker; Broker; warehouses, including manufacturers` and distributors` warehouses, chain warehouses for pharmaceuticals and wholesale warehouses for pharmaceuticals; independent wholesalers of medicines; and retail pharmacies engaged in wholesale distribution. (iii) Reporting. The requirements of point (c)(1)(i) and (c)(1)(ii) of this Section relating to the submission of post-market alerts within 15 days shall also apply to any person whose name appears on the label of an authorised organic product as manufacturer, packager, distributor, common manufacturer, joint manufacturer or any other participant involved in shared manufacturing. In order to avoid unnecessary duplication of work in submitting reports to the FDA required by paragraphs (c)(1)(i) and (c)(1)(ii) of this Article, the obligations of persons other than the applicant for the final biologic product may be fulfilled by providing the applicant for the final product with any serious adverse experience reports. If an individual chooses to submit adverse experience reports to the applicant rather than to the FDA, the individual must provide each report to the applicant appropriately within 5 calendar days of the first receipt of the information by the individual, and the applicant must then comply with the requirements of this section.
法律相談会のご案内


桜上水法律事務所のご案内
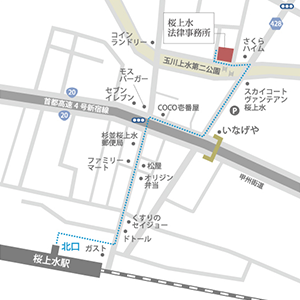
【所在地】
〒168-0073
東京都杉並区下高井戸3丁目11番1号
(京王線桜上水駅北口徒歩3分)
【電話番号】
03-6379-7810
【FAX番号】
03-6379-7828
【執務時間】
9:30~17:30(月~金)